The following references, which are the sources for most of the current module's figures, provide a tutorial on protein structures and an excellent hyperlinked glossary of genetic terms.
·
"Protein Structure Basics" by Bernhard Rupp, UCRL-MI-125269, Lawrence Livermore National Laboratory, 2000, http://ruppweb.dyndns.org/·
"Talking Glossary of Genetic Terms," National Human Genome Research Institute, http://www.nhgri.nih.gov/DIR/VIP/Glossary
In the module "Computational Science and Web-Accessed Databases," we introduced a "Genomic Example" involving web-accessed genomic databases. The current module provides the biological background for understanding such databases and a discussion of their characteristics. The next module on "Genomic Sequence Comparison" presents the dynamic programming technique for measuring the similarity of two DNA sequences, and the module "Searching Genomic Databases" considers algorithms for discovering regions of sequence alignment. Thus, with our knowledge HTML, databases, and CGI programming, we will be able to develop form-based web pages and CGI programs for interfacing between web pages and genomic databases, for processing DNA sequences, and for returning results to the user. Such applications fall in the domain of bioinformatics.
A newly developing area of computational science, called
bioinformatics, deals with the organization of biological data, such as
in databases, and the analysis of such data. Recently, enormous strides have been
made in genetics, due in part to the power of bioinformatics, as the following examples illustrate:
| Geneticists in Sweden and with Merck Research Laboratories in West Point, Pennsylvania, studied families with a high occurrence of macular degeneration, which causes loss of vision in old age. "Once the combined team got to within 800,000 bases of it [a section of the chromosome], the researchers searched computer databases for potential genes in that target region. Both labs then looked for mutations in those genes in family members with the disease but not in people with normal vision." One of the groups "found that the mutations consistently appeared" in a particular gene. The researchers are now investigating how mutations in this gene initiate the pathology. ["New Gene Found for Inherited macular Degeneration", Human Genetics News Focus column by Elizabeth Pennisi, v. 281, 3 July 1998, Science, p. 31] |
| A microbial ribosomal DNA (rDNA) sequence database is particularly valuable because biologists have not been able to culture many bacteria and viruses outside the body. By extracting the rDNA and comparing its genetic makeup with data from the database, scientists have been able to determine organisms that cause Crohn's disease, which is an inflammatory bowel disorder, and several other diseases ["The Search for Unrecognized Pathogens" by David A. Relman, v. 284, 21 May 1999, Science, pp. 1308-1310]. |
|
|
|
|
|
|
|
Alanine |
|
|
|
Cysteine |
|
|
|
Aspartic Acid |
|
|
|
Glutamic Acid |
|
|
|
Phenylalanine |
|
|
|
Glycine |
|
|
|
Histidine |
|
|
|
Isoleucine |
|
|
|
Lysine |
|
|
|
Leucine |
|
|
|
Methionine |
|
|
|
Asparagine |
|
|
|
Proline |
|
|
|
Glutamine |
|
|
|
Arginine |
|
|
|
Serine |
|
|
|
Threonine |
|
|
|
Valine |
|
|
|
Tryptophan |
|
|
|
Tyrosine |
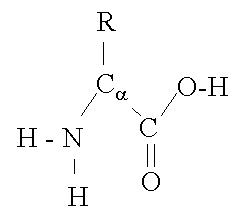
Figure 1 Structure of an amino acid ["Protein Structure Basics" by Bernhard Rupp, UCRL-MI-125269, Lawrence Livermore National Laboratory, 2000, http://ruppweb.dyndns.org/Xray/tutorial/protein_structure.htm]
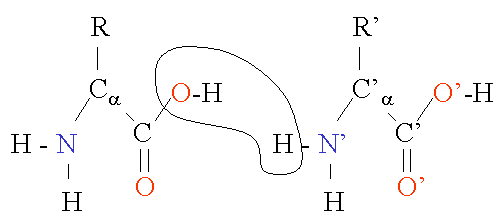
Figure 2 Chain of two amino acids ["Protein Structure Basics" by Bernhard Rupp, UCRL-MI-125269, Lawrence Livermore National Laboratory, 2000, http://ruppweb.dyndns.org/Xray/tutorial/protein_structure.htm]
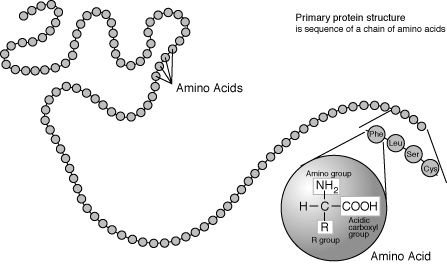
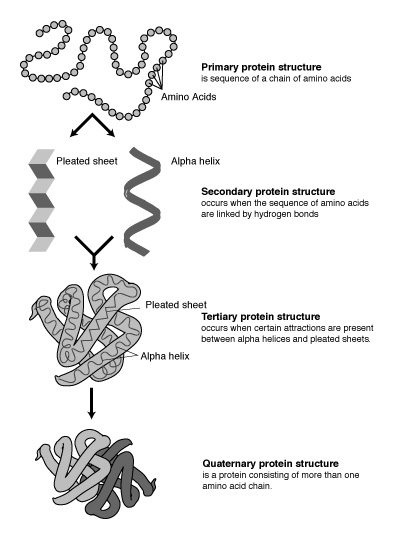
The linear sequence of residues is called the primary
structure of a protein (see Figure 3). However, interactions between
hydrogens and oxygens of the amino and carboxyl groups of different amino acids
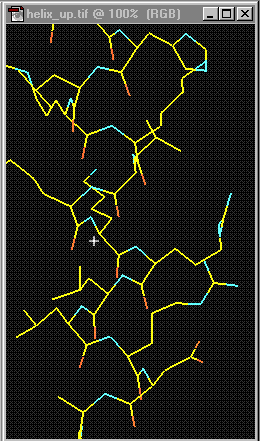
may result in regular arrangements, called the secondary structure of the protein.
For example, a helix is one type of secondary structure (see Figure
5). Chemical interactions that take place between R-groups of nonadjacent
amino acids maintain this structure. The primary structure governs these higher
order structures of proteins, which are essential in determining the
three-dimensional conformation of the protein. A single polymer of amino acids
might be more properly called a polypeptide. Some functional proteins are
made up of only one polypeptide, whereas many proteins are made up of more than
one polypeptide. For instance, hemoglobin consists of two pairs. Proteins that
consist of two or more interacting polypeptides are said to exhibit quaternary
structure (see Figure 5). These higher-order structures are very important
because they determine the overall shape of the protein, possible bindings of the protein, and, thus, its function.
Figure 3 Primary protein structure ["Talking Glossary of Genetic Terms," National Human Genome Research Institute, http://www.genome.gov/Pages/Hyperion//DIR/VIP/Glossary/Illustration/amino_acid.shtml]
Figure 4 Helix ["Protein Structure Basics" by Bernhard Rupp, UCRL-MI-125269, Lawrence Livermore National Laboratory, 2000, http://ruppweb.dyndns.org/Xray/tutorial/protein_structure.htm]
Figure 5 Protein structures ["Talking Glossary of Genetic Terms," National Human Genome Research Institute, http://www.genome.gov/Pages/Hyperion//DIR/VIP/Glossary/Illustration/protein.shtml]
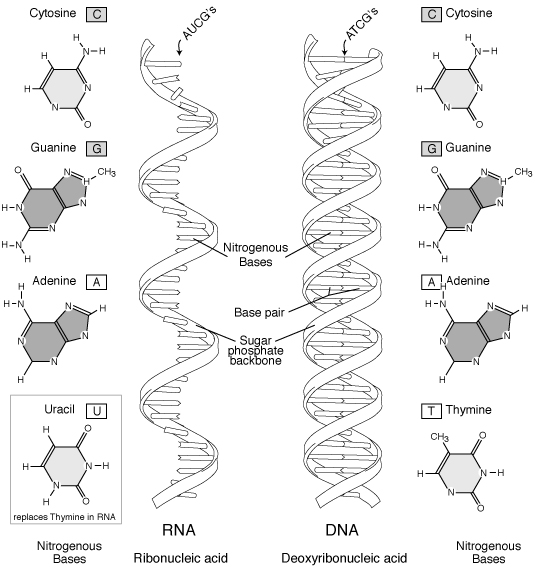
Figure 6 RNA and DNA ["Talking Glossary of Genetic Terms," National Human Genome Research Institute, http://www.genome.gov/Pages/Hyperion//DIR/VIP/Glossary/Illustration/rna2.shtml]
Bases in one strand may bond with bases in another. Because of their structure,
A and T always bond together, and C and G always bond together. Each pair
is said to be made up of complementary bases and is referred to as a base pair (bp).
The number of such base pairs is used to describe the length of a DNA
molecule. Because of pairing consistency, by knowing the sequence of
bases in one strand, we can deduce the sequence of bases in the other strand through
reverse complementation. For example, suppose one sequence is s
= ATGAC. Because of the required pairing, A - T and C - G, we know the base pairs
must appear as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A | T | G | A | C |
|
|
C | A | G | T | A |
|
|
G | T | C | A | T |
| s: | G | T | A | C | C | T |
| s’ | T | C | C | A | T | G |
|
|
A | G | G | T | A | C |
In contrast to DNA, RNA (ribonucleic acid) is a single strand
of nucleotides made up of ribose sugars and bases A, C, G, and U instead of the
nitrogen base thymine (T) (see Table 2). Several types of RNA with different functions exist in the
cell.
Table 2. Bases in DNA and RNA
|
|
|
|
|
|
| adenine |
|
|
|
|
| guanine |
|
|
|
|
| cytosine |
|
|
|
|
| thymine |
|
|
|
|
| uraciler |
|
|
|
|
Figure 7 Gene as contiguous section of chromosome [Figure slightly modified from graphics at "Talking Glossary of Genetic Terms," National Human Genome Research Institute, http://www.genome.gov/Pages/Hyperion//DIR/VIP/Glossary/Illustration/gene.shtml]
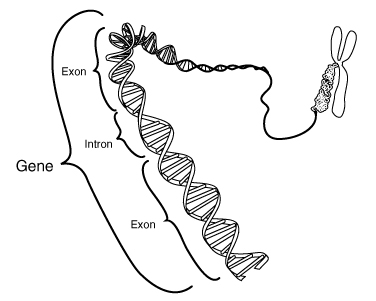
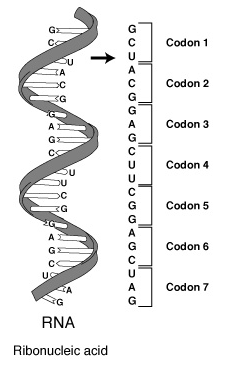
For simplicity, we assume that a particular protein in an organism corresponds to exactly one gene. In a gene, a sequence of three nucleotides (triplet) specifies an amino acid. For example, the sequence ACG or the codon ACA encodes the information for the amino acid Threonine (Thr). The genetic code represents such a correspondence between these triplets and the amino acids they specify. With four base choices, a pair of bases could only encode information for (4)(4) = 16 amino acids. With three bases, (4)(4)(4) = 64 possible triplets exist. Several, such as ACG and ACA, encode the same amino acid; and three sequences do not encode for any amino acid.
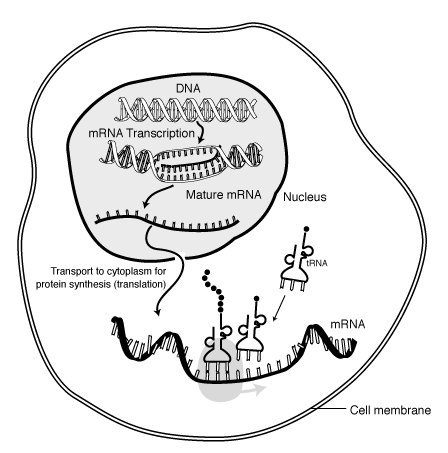
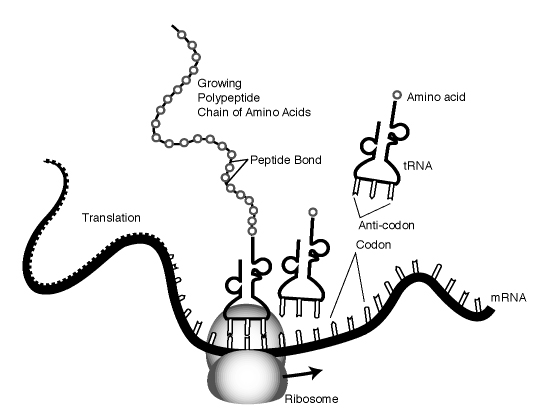
Protein synthesis is the process of using genetic code to direct the building of proteins. Synthesis begins in the nucleus, where enzymes catalyze the production of a molecule of RNA, termed messenger RNA or mRNA. As Figure 8 illustrates, each DNA triplet specifies a complementary sequence of three nucleotides, which we call a codon, in the RNA. The synthesis of RNA is called transcription. During transcription, base pairing ensures formation of a strand of RNA that is complementary to the gene sequence with U replacing T.
Codons in RNA ["Talking Glossary of Genetic Terms," National Human Genome Research Institute, http://www.genome.gov/Pages/Hyperion//DIR/VIP/Glossary/Illustration/codon.shtml]
As Figure 9 shows, transcription represents only the first step in protein synthesis. The initial transcript must be processed through a complex process of chemical changes that includes removal of portions of the RNA and splicing back together of loose ends. However, this modified mRNA molecule is in the nucleus with a double thickness of membrane separating the nucleus from the cytoplasm, or area surrounding the cell nucleus; while protein synthesis must conclude in the cytoplasm on small structures called ribosomes. The mRNA molecule must be transported from the nucleus into the cytoplasm before any protein can be synthesized.
Figure 9 Protein synthesis ["Talking Glossary of Genetic Terms," National Human Genome Research Institute, http://www.genome.gov/Pages/Hyperion//DIR/VIP/Glossary/Illustration/mrna.shtml]
After movement to the cytoplasm, the mRNA attaches to a ribosome, which essentially reads the code (sequence of codons) specified by the gene. The ribosome brings together the mRNA strand with various molecules of another type of very small RNA, called transfer RNA (tRNA). There are many different tRNA molecules, each of which attaches very specifically to only one type of amino acid. The ribosomal enzymes must ensure that each codon of the mRNA combines with a tRNA that carries the correct amino acid in the nascent protein sequence. This process is possible because each tRNA molecule contains a triplet code, called an anticodon. The anticodon then base pairs with the complementary codon of the mRNA, ensuring addition of the correct amino acid. The ribosome moves down the mRNA molecule one codon at a time, allowing the correct tRNA with the specified amino acid to base pair. Enzymes of the ribosome also help by catalyzing the formation of peptide bonds between the amino acid and the growing peptide chain (see Figure 10). Eventually, the ribosome reaches a codon that does not code for any amino acid, which signals the ribosome to stop. Voila! With the help of some RNA and ribosomes, we have a protein with an amino acid sequence that the DNA sequence of the gene specified.
Growing peptide chain ["Talking Glossary of Genetic Terms," National Human Genome Research Institute, http://www.genome.gov/Pages/Hyperion//DIR/VIP/Glossary/Illustration/peptide.shtml]
The National Center for Biotechnology Information (NCBI), USA, maintains GenBank, a database of more than 920,000 plant and animal DNA sequences from over 16,000 species. The database has been doubling about every 18 months. Figure 11 is an example of a GenBank record. The accession number, here M82814, is a key and field identifiers describe the contents of the fields, which are strings. One can search the database by keyword or sequence. Other databases include DNA database EMBL, protein sequence database PIR, PDB database for three-dimensional structures of proteins, and EcoCyc Encyclopedia of Escherichia coli Genes and Metabolism.
Figure 11. A GenBank record (Click here)
A high degree of variability is another aspect of the
complexity of biological data. Often there are exceptions, or new discoveries
cause revisions or additions to possible values. For example, besides the 20 common
amino acids, we use three other symbols for ambiguous or unknown situations. Thus,
data types must be flexible with few constraints.
Because of this variability, schemas change rapidly. Database
designers must manage extensions to the schemas as scientific knowledge expands.
Many genomic databases are re-released annually or semiannually.
To add to the complexity involving data types and schemas,
two biologists or two databases may represent the same data in different ways.
However, scientists need to be able to compare their findings.
One of the most significant access characteristics of
genomic databases is that scientists read, or query, such databases extensively
but rarely write to one. For example, each month in 2000, the MITOMAP database
had over
10,000 people that query the database but fewer than five who made additions to
it.
As another characteristic, these querying users have a
limited knowledge of the database schema design. Hence, the web access to the
database must be flexible and intuitive to handle a variety of queries. Often,
these queries are complex and involve multiple data sets.
Moreover, biologists are interested in the data in context.
For example, a biologist might want to know similar sequences to a query sequence,
where they are alike, where they are different, how the sequences compare, functions
of similar subsequences, etc. By making such comparisons, he or she hopes to deduce
functions of the query sequence.
Biologists must query the most recent version of a database
but also must be able to access earlier versions to examine data and repeat procedures.
Thus, a genomic database must provide access to historic data.
The following is a summary of the main characteristics
of genomic databases:
a. Find its complementary strand.2. Repeat Exercise 1 for the sequence CTGGATAGGCCAGT.
b. Find the corresponding sequence of bases in mRNA.
c. Give 6 possible sequences of codons for the protein it produces.
a. The information on Accession Number M82606. Hint: You can use the online search at the GenBank site.4. For the GenBank entity record in Figure 11, determine possible attributes and their types.
b. The disease-causing agent Vibrio cholerae, which causes cholera. Search the vital sequence section of the database.
Copyright © 2002, Dr. Angela B. Shiflet
All rights reserved