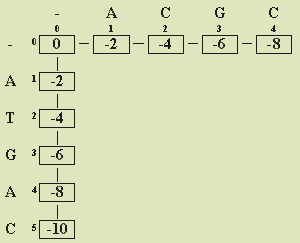Sequence comparison to determine how similar or different
two sequences are is a fundamental operation of computational biology. Perhaps
two laboratories want to compare their work on the same gene; or a biologist might
want to determine similarities in gene sequences from different organisms, such
as yeast and mouse. Programs that recreate a large DNA sequence from fragments
use sequence comparison. As another example, some biologists are interested in
searching databases for sequences that are locally similar to a given sequence
in an attempt to discover gene function. (Refer to the "Genomic
Data" module for background material for the current module.)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For example, with indicated column scores, the alignment
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| column scores: | 1 | -2 | -1 | -1 | 1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| column scores: | -2 | -2 | -2 | 1 | 1 | -2 | -2 |
Because we obtain the same column scores regardless of which sequence is in
the first row, the order in which we write the sequences is irrelevant for
determining an alignment score. Thus, the similarity of two sequences does
not depend on which we write first. For example, the similarity of ATGAC
and ACGC is the same as the similarity of ACGC and ATCAC.
|
|
|
|
Figure 1 Initial values in similarity matrix
Array position a[i][j]
is the similarity score for prefixes s[0..i-1] and t[0..j-1].
For example, a[3][2] gives the similarity score for s[0..2] (ATG
in our example) and t[0..1] (AC here). In row 0, we write the on-going scores for matching all spaces with prefixes
of t. For example, aligning a space with the prefix t[0..0] = A
costs -2; two spaces corresponding to t[0..1] = AT has a cumulative value
of (-2) + (-2) = -4; three spaces and ACG, adds -2 to the previous score to obtain
(-4) + (-2) = -6; and four spaces with t yields (-6) + (-2) = -8. Symmetrically,
in column 0, we place the on-going scores for matching prefixes of s
with all spaces. We draw line segments between subsequent values to indicate the
derivation steps.
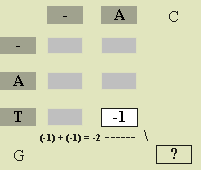
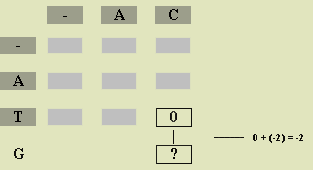
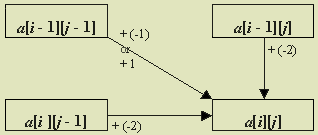
We give a general formula for score a[i][j] below, but first we illustrate the process with a particular example. To determine
a[3][2], we only must know the row 3 heading (G), column 2 heading
(C), and the scores in positions above (a[2][2]), to the left (a[3][1]),
and diagonally left above (a[2][1]) the desired position. Figure
2 illustrates this situation. We obtain the best alignment of ATG and AC in
one of three ways:

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
To summarize, a[3][2] is the maximum of the following
values:
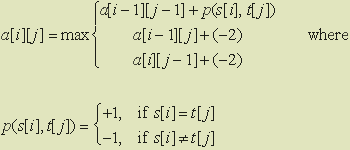
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
| _ | A | C | G | C | ||||||
| 0 | 1 | 2 | 3 | 4 | ||||||
| _ | 0 |
0 |
- |
-2 |
- |
-4 |
- |
-6 |
- |
-8 |
| | | \ | |||||||||
| A | 1 |
-2 |
1 |
- |
-1 |
- |
-3 |
- |
-5 |
|
| | | | | \ | \ | |||||||
| T | 2 |
-4 |
-1 |
0 |
- |
-2 |
- |
-2 |
||
| | | | | \ | | | \ | \ | |||||
| G | 3 |
-6 |
-3 |
-2 |
1 |
- |
-1 |
|||
| | | \ | | | \ | | | | | \ | ||||
| A | 4 |
-8 |
-5 |
-4 |
-1 |
0 |
||||
| | | | | \ | | | \ | ||||||
| C | 5 |
-10 |
-7 |
-4 |
-3 |
0 |
Figure 5 displays the similarity matrix of Figure 4 with the path from a[5][4] to a[0][0] indicating the best alignment. Using Figure 5, Figure 6 gives the optimum alignment of ATGAC and ACGC. Sometimes, more than one alignment yields the same similarity.
Figure 5. From Figure 4, path indicating best alignment of ATGAC and ACGC in bold
| _ | A | C | G | C | ||||||
| 0 | 1 | 2 | 3 | 4 | ||||||
| _ | 0 |
0 |
- |
-2 |
- |
-4 |
- |
-6 |
- |
-8 |
| | | \ | |||||||||
| A | 1 |
-2 |
1 |
- |
-1 |
- |
-3 |
- |
-5 |
|
| | | | | \ | \ | |||||||
| T | 2 |
-4 |
-1 |
0 |
- |
-2 |
- |
-2 |
||
| | | | | \ | | | \ | \ | |||||
| G | 3 |
-6 |
-3 |
-2 |
1 |
- |
-1 |
|||
| | | \ | | | \ | | | | | \ | ||||
| A | 4 |
-8 |
-5 |
-4 |
-1 |
0 |
||||
| | | | | \ | | | \ | ||||||
| C | 5 |
-10 |
-7 |
-4 |
-3 |
0 |
Figure 6. Optimal alignment of ATGAC and ACGC obtained from Figure 5
|
|
|
|
|
|
|
|
|
|
|
|
|
|
score(s, t, match Value, mismatch Value, spacePenalty, a) Procedure to determine array of similarity values for two sequences of bases Pre: s and t are non-empty sequences of bases. match Value is the numeric value for two paired bases in an alignment agreeing. mismatch Value is the numeric value for two paired bases in an alignment disagreeing. spacePenalty is the numeric value for a base being paired with a space in an alignment. match Value > mismatch Value match Value > spacePenalty a is an m-by-n array, where m is the length of s plus 1 and n is the length of t plus 1. Post: a is an array of similarity values for prefixes of s and t. The value in the bottom, right corner is the similarity of s and t. Algorithm: m <-- length of s n <-- length of t for i from 0 through m, do the following: a[i][0] <-- i * spacePenalty for j from 0 through n, do the following: a[0][j] <-- j * spacePenalty for i from 1 through m, do the following: for j from 1 through n, do the following: if (s[i - 1] equals t[j - 1]) diagonalValue <-- match Value else diagonalValue <-- mismatch Value a[i][j] <-- max(a[i - 1][j - 1] + diagonalValue, a[i - 1][j] + spacePenalty, a[i][j - 1] + spacePenalty) |
align(s, t, a, match Value, mismatch Value, spacePenalty, sAlignment,
tAlignment)
Algorithm to determine optimal alignment of two sequences of bases
Pre:
s and t are sequences of bases.
a is the array of similarity values for prefixes of s and t, as completed by score.
spacePenalty is the numeric value for a base being paired with a space in an alignment.
Post:
sAlignment and tAlignment are sequences in the optimal alignment for
sequences s and t, respectively, with a dash indicating a space.
Algorithm:
sLength <-- length of s
tLength <-- length of t
recAlign(s, t, a, match Value, mismatch Value, spacePenalty, sAlignment,
tAlignment, sLength, tLength)
|
recAlign(s, t, a, match Value, mismatch Value, spacePenalty, sAlignment,
tAlignment, i, j)
Algorithm to determine optimal alignment of two prefix sequences of
bases
Pre:
s and t are sequences of bases.
a is the array of similarity values for prefixes of s and t, as completed by score.
i is an index that is less than or equal to the length of s.
j is an index that is less than or equal to the length of t.
spacePenalty is the numeric value for a base being paired with a space in an alignment.
Post:
sAlignment and tAlignment are sequences in the optimal alignment for
prefixes s[0..i - 1] and t[0..j - 1], respectively, with a dash indicating
a space.
Algorithm:
if (i equals 0 and j equals 0)
sAlignment <-- null
tAlignment <-- null
else
if (i > 0 and a[i][j ] equals a[i - 1][j] + SpacePenalty // above
recAlign(s, t, a, match Value, mismatch Value, spacePenalty,
sAlignment, tAlignment, i - 1, j)
sAlignment <-- sAlignment + s[i - 1]
tAlignment <-- tAlignment + ‘-‘
else if (j > 0 and a[i][j ] equals a[i][j - 1] + spacePenalty) // left
recAlign(s, t, a, match Value, mismatch Value, spacePenalty,
sAlignment, tAlignment, i, j - 1)
sAlignment <-- sAlignment + ‘-‘
tAlignment <-- tAlignment + t[j - 1]
else // diagonal
recAlign(s, t, a, MatchValue, MismatchValue, spacePenalty,
sAlignment, tAlignment, i - 1, j - 1)
sAlignment <-- sAlignment + s[i - 1]
tAlignment <-- tAlignment + t[j - 1]
|
1. Consider the two sequences ATGAC and ACGC.
a. List every possible alignment in which ATGAC has no blanks.
b. List every possible alignment in which ATGAC has at least one blank
and ACGC has only blank(s) at the end.
2. a. Trace through the score algorithm for the strings and similarity array in
Quick Review Question 4 and Figure
4.
b. Trace through align and recAlign algorithms for the strings and similarity array in
Quick Review Question 5 and Figure
5.
3. a. Develop the similarity array for strings s = CGTA and t
= GCATG using the scoring of +1, -1, -2 from the example in this module.
b. What is the similarity score?
c. Give the optimal alignment that align returns.
d. Give all optimal alignments for these strings.
4. Repeat Exercise 1 for strings s = CGTA and t = GCATG.
5. Repeat Exercise 1 for strings s = ATGAC and t = ACGC
using the scoring that a mismatch or space costs 1, while a match has score 2.
6. What is the complexity of score?
7. What is the complexity of recAlign?
8. Change the priority of the align algorithm
so that when a choice exists, the algorithm gives priority to going vertically
up, next to going on the diagonal, and finally to going to the left. Because priority
is given to going up, the resulting alignment is called the "upmost alignment".
Give the additional parameters that align and recAlign need.
9. Change the priority of the recAalign algorithm
so that when a choice exists, the algorithm gives priority to going to the left,
next to going on the diagonal, and finally to going vertically up. Because priority
is given to going to the left, the resulting alignment is called the "downmost
alignment". Give the additional parameters that align and recAlign
need.
Copyright © 2002, Dr. Angela B.
Shiflet
All rights reserved