
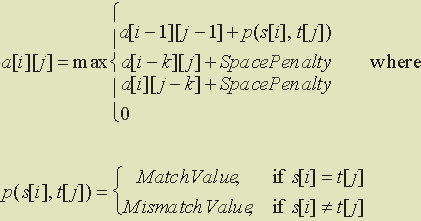
Example 1.
Using the Smith-Waterman algorithm with sequences s = CCTGAGTT and
t = ACCTAGCGA, we determine the best local alignment and its similarity
score. Suppose the space penalty is -7; the match value is +5; and the mismatch
value is -4. Assume that initial gaps are worth 0, so that we initialize
row 0 and column 0 with all zeros. Note that the letters C, T, G, and A
could represent bases or amino acids. Moreover, the same algorithm applies
to different letters.
Click here for the completed matrix. The maximum value in the table is 18. Following the line segments backward until arriving at a zero, we discover the best local alignment to be for the segment CCTGAG from s and CCT-AG from t, as follows:
|
|
|
||
|
A R N D C Q E G H I L K M F P S T W Y V B Z X
A 2
R -2 6
N 0 0 2
D 0 -1 2 4
C -2 -4 -4 -5 12
Q 0 1 1 2 -5 4
E 0 -1 1 3 -5 2 4
G 1 -3 0 1 -3 -1 0 5
H -1 2 2 1 -3 3 1 -2 6
I -1 -2 -2 -2 -2 -2 -2 -3 -2 5
L -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6
K -1 3 1 0 -5 1 0 -2 0 -2 -3 5
M -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6
F -4 -4 -4 -6 -4 -5 -5 -5 -2 1 2 -5 0 9
P 1 0 -1 -1 -3 0 -1 -1 0 -2 -3 -1 -2 -5 6
S 1 0 1 0 0 -1 0 1 -1 -1 -3 0 -2 -3 1 2
T 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 3
W -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -6 -2 -5 17
Y -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10
V 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4
B 0 -1 2 3 -4 1 2 0 1 -2 -3 1 -2 -5 -1 0 0 -5 -3 -2 2
Z 0 0 1 3 -5 3 3 -1 2 -2 -3 0 -2 -5 0 0 -1 -6 -4 -2 2 3
X 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
A R N D C Q E G H I L K M F P S T W Y V B Z X
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
||
|
|
|
|
|
|
|
|
||||
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
||
|
|
|
||
|

|
|
||
|
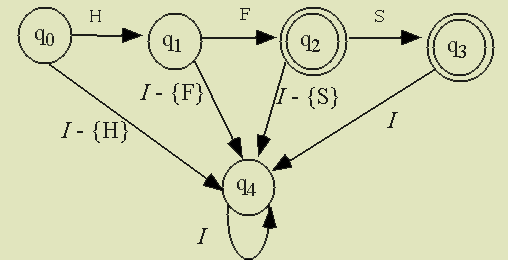
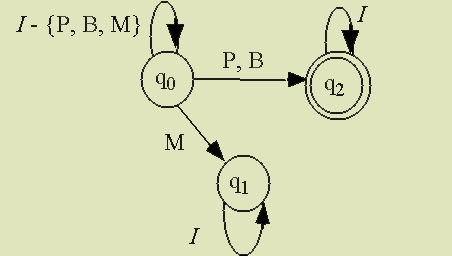
| input | N | R | S | Q | H | R | H | Q | ||
| state | q0 | q0 | q1 | q0 | q2 | q4 | q1 | q3 | q5 |
|
|
||
|
| In query: | S | D | M | C | D | R | H | Q | M | N | C | F | P | S |
| In database: | N | R | S | Q | H | R | H | Q | L | D | L | D | M | F |
| Score: | 1 | -1 | -2 | -5 | 1 | 6 | 6 | 4 | 4 | 2> | -6 | -6 | -2 | -3 |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a. Using this scheme, compute the number corresponding to PCV.13. For DNA, BLAST uses a word size of eleven bases. A typical BLAST similarity matrix uses a score of 5 for no evolution and a score of -4 for evolution. Thus, in the matrix, all diagonal elements are 5 and off-diagonal elements are -4.
b. To what index does PCV hash?
a. Write this similarity matrix.14. For DNA, a mutation between A and G or C and T, called a transition, is more likely to occur than a mutation between A and C, A and T, C and G, or G and T, called a transversion. A Transition/Transversion matrix, such as the one that follows, reflects such scoring:
b. Give the 11-mers in the DNA sequence is ATTGTAATCCATCCCTAGGTTATAC.
c. Give the score of ATGGTACTCCG relative to the first 11-mer in the DNA sequence of Part b.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Give the score of ATGGTACTCCG relative to the 11-mer
ACTGTAATCCA.
Copyright © 2002, Dr. Angela B.
Shiflet
All rights reserved