in Science

|
What's News in Science
|
Sickle Cell Disease – Relief at last?
by George Shiflet
|
Human hemoglobin (Hb) is a complex protein composed of 4 subunits, 2 α and 2 β globin subunits. Each subunit is attached to one molecule of heme, which contains oxygen-bonding iron (Figure 1). Sickle Cell Disease (SCD), or Sickle Cell Anemia (SCA), is caused by a single mutation in the gene for β globin. Because one gene from each parent codes for one of the β globin molecules, a person must receive one sickle cell gene from each parent to develop sickle cell anemia. In Figure 2 you see two parents who each have one copy of the normal gene (A) and one copy of the sickle cell gene (a) (Figure 2). Those parents are then considered carriers of the sickle cell gene and, therefore, show no signs of the disease. However, children born to those parents have a 25% chance to inherit the disease.
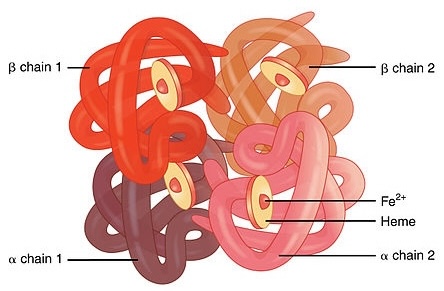
Figure 1. Diagram of human hemoglobin, showing the two α and two β globin subunits.
Copyright © by OpenStax College at https://commons.wikimedia.org/wiki/File:1904_Hemoglobin.jpg.

Figure 2. Diagram for inheritance of sickle cell disease. The parents have one copy each of the normal and sickle cell genes and are, thus, carriers of the sickle cell trait. Offspring from this pair have a 50% chance of producing children who are carriers (not shown, both types of aA combinations, where the mother could contribute a and the father an A or vice versa, therefore, 2/4 or ½ or 50% chance of a child being a carrier). There is a 25% chance of producing either a normal (AA) child or 25% probability of a child with sickle cell (aa).
Copyright © by AllGasNoBreaks at https://commons.wikimedia.org/wiki/File:Sickle_cell_disease_overdominance_and_high_fitness.jpg.
There are about 100,000 people in the United States who suffer from sickle cell disease, but there are millions of SCD sufferers worldwide. Almost all SCD individuals have ancestry from regions of the world that have endemic malarial disease. Data indicates that individuals in those areas that are carriers are more likely to have partial protection from severe malarial disease, whereas those individuals with genotype AA suffer higher malarial mortality rates. The Aa genotype has a selective advantage in those populations, and, therefore, the defective gene remains in circulation.
The single genetic variant codes for a β globin that has a very different shape than that from a normal gene, which changes drastically the shape of the hemoglobin molecule. Normal red blood cells (RBCs) are primarily membrane-bound packets containing about one quarter billion hemoglobin molecules, generating a flexible, rounded shape. Trillions of these very flexible containers can move very smoothly through all the twist and turns of the circulatory system, including the smallest vessels in that system. Sickle cell β globin tends to form fibrils of globin molecules, which deforms the RBC shape into a sickle or crescent shape (Figure 3). One of the major functions of hemoglobin is to transport oxygen from capillaries in the lungs to all the tissues of the human body. Sickle cells transport less oxygen than normal RBCs and clump with other red blood cells, blocking the blood supply to vital organs (Figure 4).
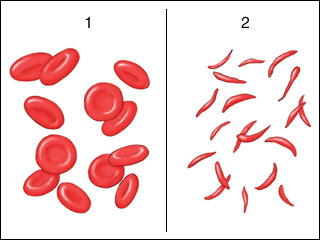
Figure 3. Normal RBC’s vs sickle shaped cells
Copyright © by Pkleong at English Wikibooks at https://commons.wikimedia.org/wiki/File:Sickle_cell_anemia.jpg.
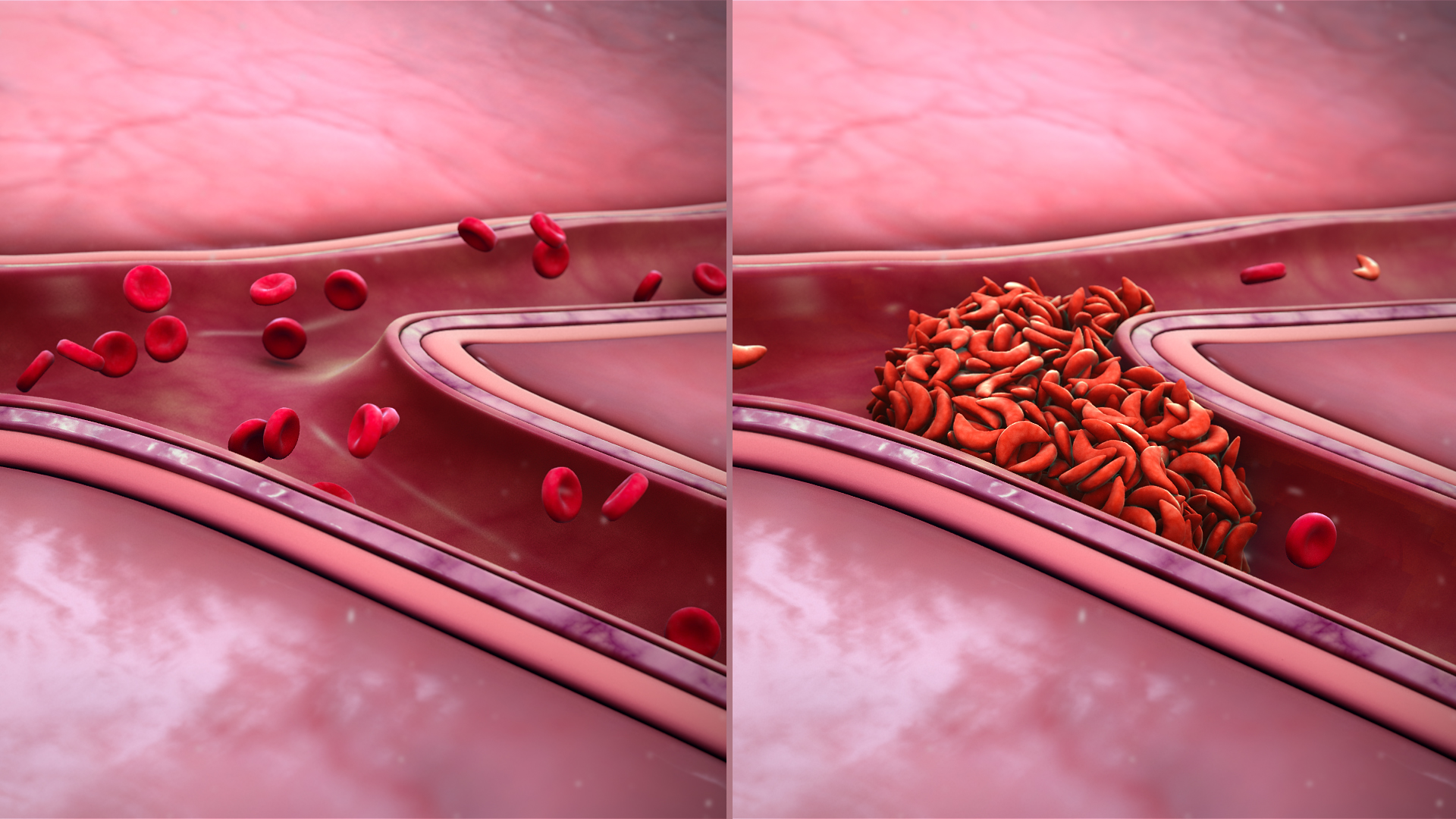
Figure 4. Comparison of normal RBC’s and sickled RBC’s in blood vessels. Note tendency of the sickled form to form clumps.
Copyright © by ShareAlike 4.0 International http://www.scientificanimations.com/wiki-images/.
Table. Major symptoms and possible complications in sickle cell disease
|
anemia |
abnormal RBC’s short-lived or destroyed in spleen → shortage of healthy RBC’s reduces oxygen → shortness of breath, fatigue, dizziness |
|
intense pain & swelling |
brought on by blockages by clumps of deformed RBC’s; frequently affects extremities (arms/hands/fingers; legs/feet/toes), chest. Blockage to deep veins of legs or to the lungs can promote blood clots |
|
yellowish color skin/eyes |
break up (hemolysis) of large numbers of fragile RBC’s |
|
organ damage |
liver, gall bladder, kidney, spleen, lungs, eyes; liver damage |
|
vision problems; blindness |
reduced oxygen and nutrients damages retina → blindness |
|
increased susceptibility to infection |
enlargement and damage to the spleen impairs body’s ability to fight infection; blockage of very small vessels of the lungs → similar to symptoms of pneumonia (fever, aches, severe cough) |
|
impaired growth, puberty |
shortage of healthy RBC’s → lack of sufficient nutrients and oxygen |
|
complications to pregnancy |
increased risk of pre-eclampsia (high blood pressure), blood clots, pre-term delivery |
|
stroke |
blockage of blood supply to the brain |
Relief from a Surprising Source
Did you know that bacteria are susceptible to viral infections, just like we are? Bacterial viruses are called phages, and they are very common. All viruses are rather selfish, only seeking havens for their own replication, oblivious to the havoc they generate for their host cells. Exposure to a specific type of phage will leave random pieces of the viral DNA, which a bacterium can incorporate into its own DNA. These pieces of viral DNA are inserted into the bacterial DNA in specific patterns, forming CRISPR (Clustered Regularly Interspersed Short Palindromic Repeats) formations, or arrays. These CRISPR regions produce specific RNA segments (gRNA) that can bind to the viral DNA. When bound, the guide RNA, or gRNA, earmarks the viral DNA for a bacterial enzyme, called Cas9, which destroys the viral DNA. Consequently, this Cas9 apparatus acts to immunize the bacterial cell against the same phage type or a closely related phage type. Scientist have already borrowed the CRISPR system to relieve human patients of Sickle Cell Disease.
We have known for some time the cause of sickle cell disease, but up to now we have mostly succeeded in managing its symptoms and complications (e.g., transfusions, etc.). One cure employing a bone-marrow transplant has many complexities.
Now, CRISPR gene editing is one of several possible techniques being tested that might cure SCD as well as other genetic diseases, some without complications of bone-marrow transplants. Adapting and using the CRISPR technologies, we may correct a number of genetically caused or transmitted disease conditions. In 2019, the first patient underwent a clinical trial that applied the CRISPR technique to correct a heritable defect in the hemoglobin that causes sickle cell disease.
A medical team at Vanderbilt University removed stem cells that produce red blood cells from her marrow and concentrated them. During early development, stem cells from a human fetus produce a special hemoglobin, hemoglobin F (2 α subunits and 2 γ subunits), which transports oxygen from the mother’s bloodstream to throughout the developing fetus. After birth, hemoglobin F is mostly, although not completely, replaced by hemoglobin A. The expression of the gene for Hb F is repressed by a genetic repressor element that is activated after birth. These first treatments using CRISPR worked by disrupting the repressor DNA (e.g., making a genetic deletion), thereby reactivating the Hb F gene in the collected stem cells. The repressor DNA is the target, rather than viral DNA. After using chemotherapy to knock out the patient’s defective marrow cells, doctors transplanted into the bone marrow the concentrated stem cells, genetically modified by CRISPR. The blood cells produced by these stem cells synthesized Hb F (as opposed to the defective Hb A), which functioned properly to supply the body with oxygen to produce RBCs of normal shape. Using the CRISPR technique, the patient was cured of SCD. The CRISPR technology can also be used with the DNA repair apparatus of the host cell to add or delete DNA nucleotide segments to the target.
Following this remarkable success, in various studies doctors and scientists have used the CRISPR gene-editing technique to cure other SCD patients. On December 8, 2023, the U.S. Food and Drug Administration (FDA) approved two gene-based treatments for SCD, including the first therapy that employs CRISPR. The CRISPR technique has the promise of leading to longer, healthier, and much less painful lives for those suffering with Sickle Cell Diease as well as cures for other serious genetic diseases.
https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/symptoms-causes/syc-20355876
https://my.clevelandclinic.org/health/diseases/4579-sickle-cell-anemia
https://www.seattlechildrens.org/conditions/sickle-cell-disease/treatment/
https://www.cdc.gov/ncbddd/sicklecell/recommendations.html
https://www.childrenshospital.org/conditions/sickle-cell-disease